El lupus eritematoso sistémico (LES) es la enfermedad autoinmune sistémica más frecuente. Presenta compromiso cardíaco en un 50-60%, y puede comprometer cualquier estructura cardíaca. La miocarditis lúpica es una enfermedad poco frecuente pero potencialmente fatal.
Historia clínica:
Mujer de 20 años, procedente de Montevideo, con antecedentes de hipotiroidismo, pre esclerosis sistémica y LES diagnosticado en 2019 con empujes recurrentes asociados a poliserositis con derrame pericárdico.\ La paciente consulta por disnea de esfuerzo CFNYHA I asociada a palpitaciones regulares y recurrentes de una semana de evolución, se destaca al examen físico un ritmo regular con frecuencia cardiaca de 150 lpm. El ECG reveló taquicardia sinusal, P y PR normales, eje eléctrico normoposicionado, QRS fino, sin trastornos en la repolarización.\ Paraclínica inicial con troponinas negativas y proBNP de 62 ng/dL, biomarcadores de actividad lúpica elevados. Se realizó ecocardiograma transtorácico en el cual se observó VI dimensiones normales,sin alteraciones sectoriales FEVI levemente disminuida 50% SLG disminuido -16.8%.\ AI de dimensiones normales.Cavidades derechas de dimensiones normales, función sistólica del VD conservada.Durante internación se inicia tratamiento del empuje de LES con bolos metilprednisolona e inmunoglobulinas. Desde el punto de vista CV persiste con palpitaciones y taquicardia sinusal por lo que se inicia betabloqueantes a dosis máximas toleradas por la paciente, sin lograr control de FC, se inicia ivabradina 7,5 mg día.Dado la clínica presentada por la paciente con descenso de FEVI y SLG con respecto a estudios previos se decide solicitar cardio RNM en vistas a valorar compromiso miocardio, la cual informa Ventrículos con volúmenes normales. FEVI levemente reducido 49%. Función del VD normal. Edema difuso basal miocárdico en T2. T1 mapping levemente aumentado.\ Con diagnóstico de miocarditis lúpica se completa tratamiento cardioprotector con IECA. Paciente con buena respuesta al tratamiento médico,asintomática en lo CV por lo que es dada de alta con controles periódicos en policlínica de cardiología.A los 6 meses del diagnóstico se realiza control persistiendo taquicardia sinusal con FC 105 lpm, se realiza cardio RNM de control que informa\ VI de dimensiones normales,FEVI conservada 60%. VD dimensiones normales.Sin evidencia de edema miocárdico
Pruebas complementarias:
Diagnóstico:
Discusión:
La presentación clínica de la miocarditis es diversa, presentando mayoritariamente síntomas leves o alteraciones transitorias electrocardiográficas. La RMC es útil para el diagnóstico en fase aguda si se cumplen al menos dos de los criterios de Lake Luis (aumento de la señal focal o difusa en las secuencias potenciadas en T2;\ b) realce precoz con gadolinio, y\ c) al menos un foco de realce tardío focal no isquémico).\ En cuanto al tratamiento en pacientes con disfunción sistólica se basa en diuréticos,IECA/ARAII y BB. La duración del tratamiento una vez recuperada la función sistólica no está definido.
#009 |
Hipertensión renovascular: Serie de casos
María Clara Aldabalde1
;
María Noel Rivero
1
;
Valentina Más
1
;
Paola Spósito
1
La enfermedad renovascular se expresa por perfusión sanguínea insuficiente en uno o ambos riñones, resultando en una activación desmedida del sistema renina-angiotensina-aldosterona. La hipertensión arterial (HA) secundaria a enfermedad renovascular es poco frecuente, representando menos de un 1%, mientras que en pacientes con HA severa asciende hasta un 35%. A continuación se presentan tres casos clínicos con HA secundaria de causa renovascular por estenosis de arteria renal.
Historia clínica:
Caso clínico 1: Mujer, 64 años. Sedentarismo, obesidad, tabaquismo. HA con cifras habituales de PA 170/70 mmHg, en tratamiento con siete fármacos. Cardiopatía isquémica, fracción de eyección del VI (FEVI) 46%. Caso clínico 2: Mujer, 78 años. Tabaquismo, dislipemia. HA con cifras habituales de PA 150/80 mmHg. En tratamiento con 6 fármacos. Enfermedad renal crónica con filtrado glomerular 24 mL/min. Caso clínico 3: Mujer, 43 años. HA con cifras habituales PA 140/70 mmHg en tratamiento con dos fármacos.
Pruebas complementarias:
Caso clínico 1: EcoDoppler de arterias renales: asimetría renal, riñón izquierdo atrófico, contornos lobulados y disminución marcada del espesor parenquimatoso. Velocidad de flujo de 47 cm/seg. Angiotomografía (angioTC): estenosis significativa del origen de la arteria ilíaca primitiva derecha. Estenosis ilíaca de 4 mm y estenosis de la arteria ilíaca externa homolateral. Riñón izquierdo atrófico. Interconsulta con Cirugía Vascular y ajuste de tratamiento médico con buenos resultados. Caso clínico 2: EcoDoppler renal: signos indirectos de estenosis proximal de arterias renales. AngioTC: oclusión del ostium de la arteria renal izquierda. Riñón izquierdo atrófico. Cirugía Vascular realiza plastia y recanalización endovascular sin éxito. Caso clínico 3: EcoDoppler renal: tercio proximal de arteria renal derecha se visualiza aumento de las velocidades de hasta 320 cm/seg, con velocidades dentro de límites habituales distalmente. AngioTC: estenosis de arteria renal derecha de 7 mm de longitud, inmediatamente posterior al origen. El diámetro mínimo medido es de 3,8 mm. En valoración por cirugía vascular, manteniendo controles de PA adecuados.
Diagnóstico:
HA de causa secundaria, de etiología renovascular.
Discusión:
Los tres casos analizados evidenciaron la aterosclerosis como etiología más frecuente de estenosis de arteria renal en pacientes con factores de riesgo cardiovascular. El tratamiento de esta enfermedad debe ser abordado en forma interdisciplinaria. Los estudios ASTRAL (Angioplasty and Stenting for Renal Artery Lesiones) y STAR (Stent Placement for Atherosclerotics Stenosis of Renal Artery), no encontraron resultados significativos en cuanto a la terapia intervencionista con respecto al tratamiento médico estándar. El ensayo clínico CORAL concluyó que la revascularización con stent de la arteria renal no proveía de un beneficio significativo en cuanto a la prevención de eventos cardiovasculares. En estos pacientes se realizó un tratamiento médico intensivo individualizado con buena respuesta. La evidencia demuestra la importancia de valorar el riesgo-beneficio del tratamiento intervencionista versus el tratamiento médico.
#011 |
COMUNICACIÓN INTERAURICULAR FAMILIAR: SINDROME DE HOLT ORAM O MANO-CORAZON
Agustín Fumeaux1
;
Victoria Briano
1
;
Juan Albistur
1
1 - Unidad Académica Cardiología-Hospital de Clínicas.
El síndrome de Holt Oram (SHO) es un trastorno genético poco frecuente que se hereda de forma autosómica dominante. Son características las deformaciones esqueléticas en miembros superiores (MMSS) asociado a cardiopatías congénitas, más comúnmente comunicación interauricular (CIA)(1).A pesar de ser un síndrome bien descrito, los casos documentados son limitados especialmente en Uruguay.
Historia clínica:
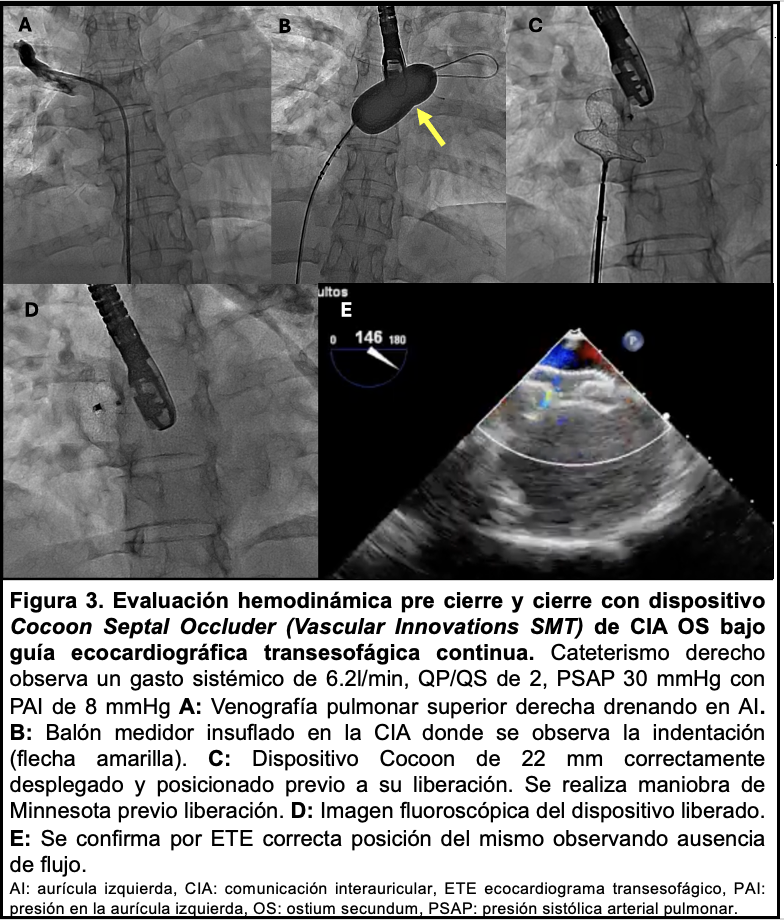
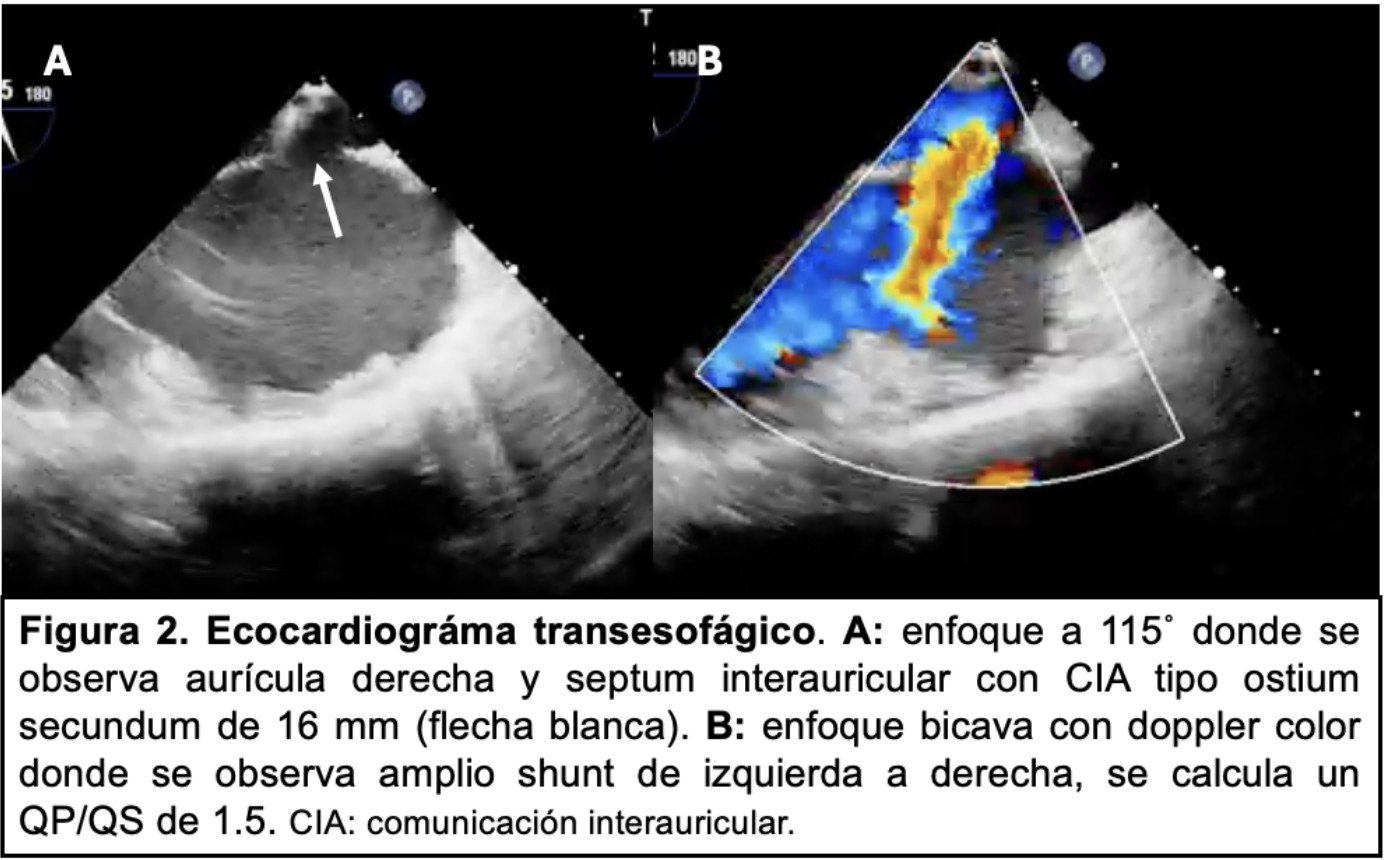
Mujer de 37 años, con antecedentes de cefalea. Antecedentes familiares de primer grado: hija requiere cierre de CIA; hijo y madre presentan alteraciones osteoarticulares de mano izquierda. Múltiples afectados en familiares de segundo grado (Figura 1A). Presenta disnea de esfuerzo de 1 año de evolución CFII y palpitaciones frente a esfuerzos. Al exámen físico cardiovascular: ritmo regular de 70 cpm, ruidos normofonéticos, soplo mesosistólico eyectivo máximo en foco pulmonar, sin elementos de falla cardíaca. ECG destaca onda P prominente en DII y trastornos inespecíficos de la repolarización. Se realizó ecocardiograma transtorácico (ETT) y posteriormente transesofágico, evidenciando FEVI 60%, cavidades derechas dilatadas, movimiento paradojal del septum y CIA amplia (Figura 2).
Pruebas complementarias:
Diagnóstico:
Síndrome de Holt Oram
Discusión:
Las CIA representan 7% de todas las anomalías cardíacas, frecuentemente asociada a otros trastornos genéticos, debiendo investigarse la existencia de familiares con CIA y asociación con Marfan, Noonan, Turner, SHO, entre otros(2). La prevalencia de SHO se estima en 1 cada 100.000 nacimientos. Las anomalías de MMSS siempre están presentes e incluyen malformaciones de los huesos del carpo, pulgares trifalángicos o ausentes, posición anormal del pulgar, hipoplasia/aplasia del radio, longitud desigual de los brazos y pronación/supinación del antebrazo. Esta afectación osteoarticular varía desde casos graves con importante impotencia funcional, hasta otros leves que pueden no detectarse fácilmente dificultando el diagnóstico sindromático, retrasando el mismo hasta el nacimiento de un familiar con afectación severa osteoarticular o hasta el desarrollo de síntomas por su cardiopatía, habitualmente a mediana edad. La baja frecuencia de este síndrome y la afectación osteoarticular leve en nuestro caso puede justificar que el personal de salud no sospechara la existencia de SHO hasta el desarrollo de disnea en la etapa adulta. Las anomalías cardíacas están presentes en el 75% de los casos, siendo más frecuentes la CIA tipo ostium secundum, en menor medida la comunicación interventricular y los trastornos de la conducción. El diagnóstico se basa en los hallazgos clínicos, antecedentes familiares y puede confirmarse mediante estudio genético. El 80% de los individuos que cumplen criterios clínicos tienen la mutación del gen TBX5 en el cromosoma 12(3). La presencia de SHO en familiares de primer grado debe obligar a descartar la presencia de cardiopatía ante un caso aparentemente sano mediante la realización de radiografía de MMSS, ECG y ETT. El seguimiento de estos casos debería ser por un equipo multidisciplinario (cardiólogos, ortopedistas y genetistas). Las indicaciones de cierre de CIA no difieren de las CIA en otro contexto(1). Un mayor conocimiento sobre este tipo de síndromes es importante para aumentar la probabilidad de un diagnóstico oportuno tanto en el caso en cuestión como en su descendencia.
#018 |
Miocardiopatía dilatada y tormenta eléctrica en deportista de alto rendimiento con mutación del Gen DSG2
Julia Tabo1
;
Camila Gurascier
1
;
Federico De La Rosa
1
;
Pedro Trujillo
1
1 - Unidad Academica de Cardiología, Hospital de Clínicas.
La miocardiopatía dilatada es la vía final común de múltiples patologías, cuya investigación etiológica tiene implicancias pronósticas y terapéuticas. Se presenta un caso clínico de miocardiopatía dilatada no isquémica que se manifiesta a forma de tormenta eléctrica y shock cardiogénico en paciente deportista de alto rendimiento, y su desafío diagnóstico.
Historia clínica:
Sexo masculino, 21 años. Futbolista de tercera división. Asmático. Consulta en Emergencia por palpitaciones que lo despiertan por la noche, disnea de reposo y sudoración profusa. Durante la evaluación presenta síncope, recuperando conciencia con FC 200 cpm, PA 80/40 mmHg. ECG (figura 1) que evidencia TVS que requiere cardioversión eléctrica a 220 J con retorno a RS, reiterando TVS en 3 oportunidades. ECG basal (figura 2) evidencia QRS de 120 ms fraccionado de forma difusa, con eje eléctrico medio desviado a derecha, alteraciones de la repolarización y EV de diferentes morfologías. Evoluciona con inestabilidad hemodinámica y shock cardiogénico, con requerimiento de apoyo inotrópico y vasopresor. Se realizó manejo antiarrítmico farmacológico con amiodarona y betabloqueante, con buena evolución posterior e implante de CDAI previo al alta.
Pruebas complementarias:
Figura 1. Figura 2. Figura 3.
Diagnóstico:
El ETT evidencia VI dilatado con HVI excéntrica severa. Hipocontractilidad global difusa, aquinesia del ápex. FEVI 15%. Biauriculomegalia severa. VD disfuncionante. IMit leve secundaria. En la evaluación etiológica se realizan rutinas sanguíneas dentro de rango normal, se descartan causas tóxicas, metabólicas, e infecciosas. CACG no evidencia lesiones ni anomalías en el origen coronario. RNM cardiaca (figura 3) revela presencia de miocardiopatía dilatada de etiología no isquémica con función sistólica del VI moderadamente disminuida, con realce tardío con gadolinio muy extenso, subendocárdico y en algunos sectores transmural del VI compromete los sectores medios a nivel anterior, lateral e inferior y todos los segmentos apicales, no correspondiéndole con un territorio vascular; así como a nivel de apex y pared libre del VD. Dada la extensión del realce tardío con gadolinio corresponde a cardiopatía con alto riesgo arrítmico. Biopsia endomiocárdica destaca elementos inflamatorios crónicos inespecíficos. Panel genético identifica variante patogénica del gen DSG2, la cual no ha sido reportada previamente, así también como la variante en heterocigosis del gen MYBPC3. Se realiza mapeo con navegador que evidencia extensa fibrosis del VI, no reproduciendo arritmia, realizando ablación por radiofrecuencia para homogeneización de sustrato.
Discusión:
La miocardiopatía dilatada no isquémica se caracteriza por la dilatación ventricular y disfunción sistólica, sin que puedan atribuirse a sobrecarga hemodinámica o enfermedad arterial coronaria. La RMC es fundamental para su diagnóstico y pronóstico, destacando el realce tardío con gadolinio y el análisis genético como elementos clave en la estratificación del riesgo. Este realce tardío de gadolinio se ha asociado con un mayor riesgo de mortalidad y eventos arrítmicos, especialmente en patrones múltiples de fibrosis. Un 50% de los casos tienen un componente hereditario, estando vinculada la mutación del gen DSG2 a la miocardiopatía arritmogénica del VD. Surge la interrogante si este caso pudiera corresponder a una forma severa de presentación de la enfermedad con compromiso ventricular izquierdo, y si la asociación de mutaciones genéticas podría explicar la presentación fenotípica.
#026 |
Trombectomía mecánica en el ACV isquémico post CACG
El IAMCEST es una importante causa de morbi-mortalidad a nivel mundial. Las tasas de mortalidad han disminuido significativamente con el advenimiento de las técnicas de reperfusión temprana (farmacológicas e intervencionismo coronario percutáneo). Paradójicamente se ha observado un incremento en las tasas de ACV post intervencionismo coronario, siendo cercanas al 0.56%.En cuanto al manejo de esta complicación, la decisión debe ser individualizada optando por trombólisis farmacológica o trombectomía mecánica.
Historia clínica:
Sexo masculino, 55 años, con antecedente de tabaquismo intenso y escasos controles en salud. Sin historia cardiovascular previa.Consulta por dolor torácico opresivo con irradiación a miembro superior izquierdo y síndrome neurovegetativo acompañante. ECG evidencia elevación del segmento ST compatible con IAMCEST inferoposterior. Durante la asistencia presenta PCR en fibrilación ventricular, se realiza 1 desfibrilación retomando circulación espontánea en ritmo sinusal. Es derivado a centro de hemodinamia para CACG de emergencia.En CACG se identifica oclusión trombótica aguda de ACD proximal. Se realiza tromboaspiración y ATC1 con implante de stent liberador de drogas en ACD (Figura 1).Durante CACG presentó episodios de fibrilación ventricular, siendo revertidos con desfibrilación. Buena evolución inicial. A las 36 hs de CACG instala síndrome focal neurológico dado por afasia de expresión. Sin arritmias ni sintomatología cardiovascular asociada. Se activa protocolo de ACV.
Pruebas complementarias:
CACG (Figura 1).AngioTAC evidencia oclusión en porción M1 luego de la emergencia de la arteria temporal izquierda (Figura 2).
Diagnóstico:
Se realiza diagnóstico de ACV isquémico en paciente con intervención coronaria reciente, con uso de antiagregantes/anticoagulantes por lo que se excluye para tratamiento trombolítico. Además, su puntaje NIHHS <6 lo excluye de cobertura para trombectomía mecánica según normativa vigente del Fondo Nacional de Recursos. Dado el impacto funcional de la sintomatología (afasia de expresión), en un paciente joven, se gestiona autorización excepcional para tratamiento endovascular.A las 6 hs de iniciados los síntomas se realiza arteriografía cerebral que confirma la presencia de trombo suboclusivo en M1 con hipoperfusión distal. Se procede a trombectomía mecánica mediante aspiración, logrando extracción del trombo y mejoría en la perfusión (Figura 3).Posteriormente, el paciente presenta una recuperación significativa.A las 48 horas, se mantiene estable sin nuevos eventos cardiovasculares ni neurológicos. En control ambulatorio permanece asintomático, sin secuelas neurológicas y con buena adherencia al tratamiento de prevención secundaria.
Discusión:
Este caso resalta la creciente incidencia de ACV post intervencionismo coronario percutáneo, atribuido a factores de riesgo del paciente y del procedimiento. Se ha identificado que la edad avanzada, FA, tromboaspiración durante CACG y el uso de dispositivos mecánicos aumentan este riesgo. La mayoría de los ACV ocurren en las primeras 24-48 horas.El manejo del ACV en este contexto presenta desafíos debido a la necesidad de equilibrar el riesgo trombótico y hemorrágico del paciente.Aunque la trombólisis intravenosa es la opción estándar, la trombectomía mecánica ha demostrado ser una alternativa efectiva.Este caso enfatiza la importancia de un enfoque individualizado en la selección de terapias de reperfusión en ACV post intervencionismo coronario percutáneo y la necesidad de flexibilidad en los protocolos de tratamiento para evitar secuelas neurológicas discapacitantes.
#029 |
Linfoma de Hodgkin con Nefropatía IgA y Miocardiopatía de Etiología No Aclarada
Soledad Murguía
1
;
Rodrigo Aranco1
;
Virginia Estragó
1
1 - Unidad Académica Cardiología, Facultad de Medicina, Hospital de Clínicas..
El linfoma de Hodgkin (LH) es una entidad hemato-oncológica potencialmente curable. Las comorbilidades pueden limitar los resultados terapéuticos determinando el pronóstico. Presentamos un paciente con LH, asociado a nefropatía IgA y miocardiopatía de etiología no aclarada, que supuso un desafío diagnóstico y terapéutico.
Historia clínica:
Hombre, 43 años con FRCV (HA tratado con valsartán, dislipidemia sin tratamiento). Sin antecedentes familiares CV a destacar. IAM previo sin lesiones coronarias obstructivas (MINOCA). Migraña automedicada con AINES y ergotamínicos. Portador de LH variante esclerosis nodular Bulky Estadio III. Es valorado por cardio-oncología para iniciar tratamiento con doxorrubicina, bleomicina, vinblastina, dacarbazina. Asintomático en lo CV. Del examen físico destaca: poliadenomegalias, latido apexiano desplazado, choque amplio y sostenido, sin soplos, no edema de MM
Pruebas complementarias:
Evaluación imagenológica multimodal:- ETT. VI dilatado (DDVI: 61 mm) HVI excéntrica, FEVI: 41%, dilatación leve de aurícula izquierda e IMi leve (Figura 1).- Biomarcadores elevados (TropI: 86 ng/L, pro-BNP: 841 pg/mL).- RNM cardíaca. VI dilatado, HVI excéntrica, aquinesia apical, hipoquinesia inferolateral basal y media, FEVI: 43%, dilatación leve de AI. Ausencia de edema. Realce tardío (RT) intramiocárdico no isquémico (Figura 2).Evaluación renal:- Creatinina: 3.22 mg/dL (2021 0.9 mg/dL), Urea: 150 g/L, K: 4,7 mEq/L. - Proteinuria subnefrótica (2.8 g/L), microhematuria y normoalbuminuria.- Biopsia renal: nefropatía IgA.
Diagnóstico:
Linfoma de Hodgkin variante esclerosis nodular Bulky Estadio III.Miocardiopatía de etiología no aclarada con FEVI disminuida.Nefropatía IgA, IRA sobre ERC.
Discusión:
La presencia de cardiopatía estructural implica un alto riesgo de cardiotoxicidad, aunque la evidencia en este grupo de pacientes es limitada. La etiología de la cardiopatía puede guiar un tratamiento específico que impactará en el pronóstico CV y oncológico. La evaluación imagenológica multimodal cobra relevancia, complementando la clínica.Con un enfoque interdisciplinario, se realizó tratamiento corticoideo mejorando la enfermedad renal. Se ajustó el tratamiento hematooncológico retrasando la incorporación de antraciclinas y utilizando doxorrubicina liposomal, asociado a terapia cardioprotectora y monitoreo estrecho con ETT y biomarcadores. Tras cuatro ciclos de quimioterapia, se observó una respuesta oncológica completa, con mejoría de FEVI y disminución de biomarcadores.La etiología de la miocardiopatía sigue sin esclarecerse. La presentación clínica y los hallazgos de la RNM sugieren diversas posibles causas. Se han reportado casos de miocardiopatía infiltrativa asociada a LH, siendo una complicación rara de la enfermedad. La HVI asociada a ERC y la localización del RT sugiere enfermedad de Fabry, por lo que se solicitó dosificación de alfa galactosidasa. No presenta la progresión típica de una miocardiopatía tóxica asociada a MINOCA, y se descartó enfermedad de Chagas. La ausencia de signos de inflamación activa aleja causas autoinmunes o postinfecciosas. Está pendiente un panel genético completo para miocardiopatías genéticamente determinadas y biopsia endomiocárdica.Abordar la disfunción sistólica en pacientes oncológicos es un desafío que requiere un enfoque multidisciplinario para minimizar la toxicidad sin comprometer los potenciales beneficios del tratamiento. En este caso, las medidas cardioprotectoras permitieron completar la quimioterapia logrando la remisión completa del LH. El pronóstico vital del paciente estará determinado por la respuesta al tratamiento de su cardiopatía y ERC.
#058 |
Estenosis valvular tricuspídea asociada a electrodo de dispositivo
Rodrigo Urban1
;
Victoria Green
1
;
Martin Antelo
2
;
Diego Freire
1
La estenosis tricuspídea es una valvulopatía poco frecuente siendo la causa más común la valvulopatía reumática. Otras causas incluyen sindrome carcinoide, Lupus, tumores y defectos congénitos. Los efectos adversos de los electrodos de dispositivos sobre la válvula tricúspide son frecuentes pero determinan principalmente insuficiencia tricuspídea. La estenosis relacionada con electrodos de marcapasos es una presentación extremadamente infrecuente, habiendo no más de 15 casos publicados. La ausencia de casos determina que se consideren varias opciones terapéuticas
Historia clínica:
paciente masculino de 52 años con antecedentes de bloqueo auriculoventricular congénito a quien se le implantó un marcapasos bicameral a los 27 años. Se encontraba en estudio por una patología inmunológica no determinada. El motivo de consulta fue la aparición de edema en miembros inferiores, distensión abdominal y disnea de esfuerzo, clase funcional II. Del examen físico se destacaba: claro soplo diastólico con máxima auscultación en la región paracardíaca izquierda, gran ingurgitación yugular, edemas bilateral hasta muslos, ascitis y sindrome en menos bilateral (anasarca). La Radiografía de tórax mostró que el electrodo ventricular realizaba un bucle en su camino hacia el ventrículo derecho. Se realizó una tomografía computada toracoabdominal que informó un árbol vascular arterial sin alteraciones, dilatación y tortuosidad de la vena ácigos y venas cavas, ascitis moderada y derrame pleural bilateral en cavidad libre.
Pruebas complementarias:
Se realizó un ecocardiograma transtorácico que evidenció una fracción de eyección del ventrículo izquierdo conservada, cavidades izquierdas normales. dilatación de la aurícula derecha. Ventrículo derecho de dimensiones y función normales. A nivel de la válvula tricúspide, se observó un aumento difuso de la ecogenicidad, con un gradiente medio de 10 mmHg y un área valvular de 0.5 cm² por tiempo de hemipresión, hallazgos sugestivos de estenosis tricuspídea severa. Además, se constató dilatación fija de la vena cava inferior y de las venas suprahepáticas, derrame pericárdico moderado pleural bilateral.
Diagnóstico:
Ante este cuadro, se inició tratamiento diurético con altas dosis, logrando una evolución favorable. En conjunto con cirujano cardíaco y hemodinamistas se discutieron opciones terapéuticas percutáneas y quirúrgicas. Se decidió la realización de una intervención quirúrgica, que incluyó explante de los electrodos y el marcapasos, sustitución de la válvula tricúspide por bioprótesis, plastia de la vena cava superior e implante de marcapasos epicárdico bicameral. La cirugía se llevó a cabo con éxito. Se comprobó degeneración valvular y gran fibrosis vinculada al electrodo ventricular. No hubo complicaciones mayores en el postoperatorio inmediato.En los controles posteriores, el paciente presentó una franca mejoría del síndrome edematoso y logró un retorno progresivo a sus actividades normales en un período de cuatro meses.
Discusión:
describimos un caso muy poco frecuente de estenosis tricuspídea vinculada a electrodos de marcapasos. La clave del diagnóstico fue el soplo diastólico y los hallazgos ecocardiográficos. Las opciones terapéuticas eran la valvuloplastia percutánea de resultado incierto y con mantención de los electrodos o la quirúrgica (que se realizó en este caso), más completa y definitiva, con el inconveniente de implantar una bioprótesis en posición tricuspídea